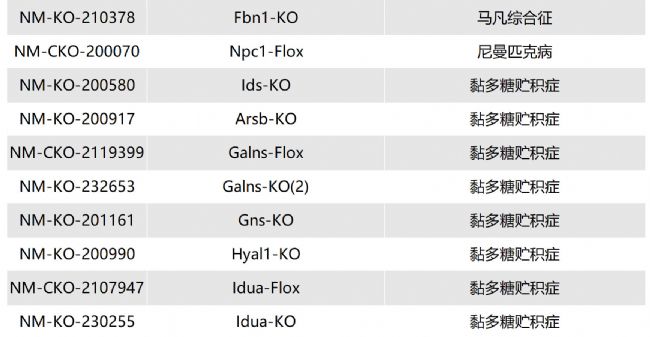
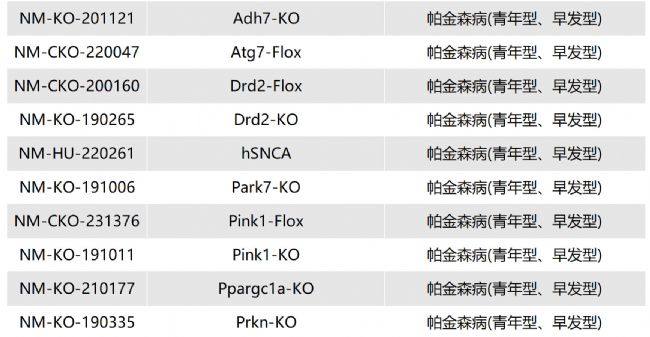
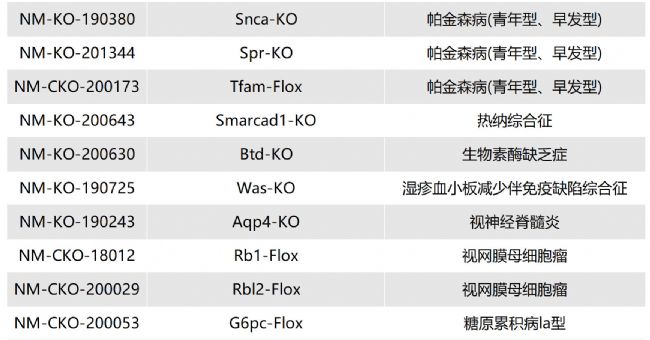
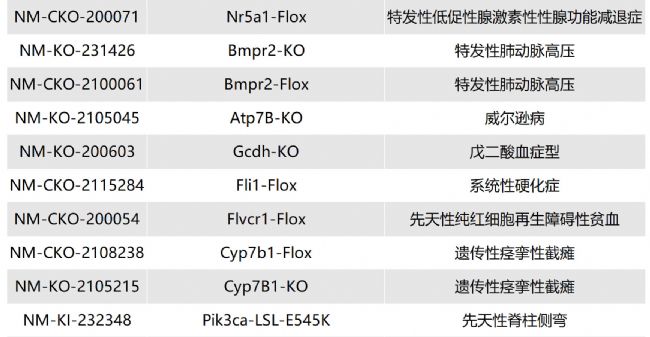
溶酶體酸性脂肪酶缺乏癥(LAL-D)小鼠模型的介紹與應(yīng)用
溶酶體酸性脂肪酶缺乏癥(lysosomal acid lipase dificiency,LAL-D)是一種罕見的、慢性的、逐漸加重的常染色體隱性遺傳性疾病。
LIPA基因會(huì)出現(xiàn)錯(cuò)義突變、無義突變、插入/缺失、剪接位點(diǎn)突變、復(fù)雜重組等,導(dǎo)致溶酶體酸性脂肪酶(lysosomal acid lipase,LAL)活性缺失或嚴(yán)重不足(<1%的正常活性)。LAL功能缺失或降低,使溶酶體降解低密度脂蛋白、膽固醇酯和三酰甘油的功能喪失或降低,膽固醇酯和三酰甘油在組織細(xì)胞內(nèi)貯積,血漿膽固醇水平升高,進(jìn)而導(dǎo)致內(nèi)臟器官出現(xiàn)黃色瘤樣變化。
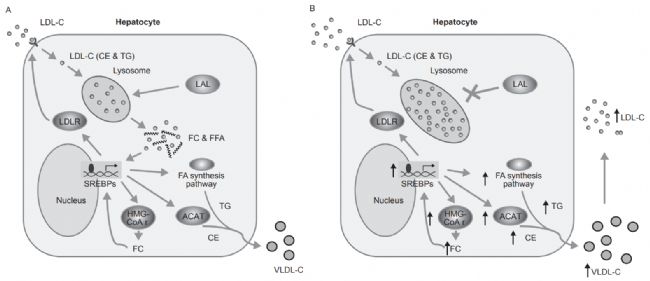
圖1. 健康個(gè)體和LAL-D患者的細(xì)胞膽固醇穩(wěn)態(tài)的示意圖[1]。
根據(jù)發(fā)病年齡和臨床表現(xiàn)不同,分為嬰兒期起病的沃爾曼病(Wolman disease,WD)和兒童及成人期起病的膽固醇酯貯積病(cholesterol ester storage disease,CESD)。目前針對(duì)溶酶體酸性脂肪酶缺乏癥的治療方法主要包括病因治療和對(duì)癥治療。Sebelipase alfa是首個(gè)獲批的酶替代治療藥物,能改善血脂異常、肝功能、胃腸道癥狀,并延長(zhǎng)生存期。
病因治療:造血干細(xì)胞移植和酶替代治療。
對(duì)癥支持治療:低脂飲食、胃腸外營(yíng)養(yǎng)、糖皮質(zhì)激素和鹽皮質(zhì)激素替代等。
南模生物L(fēng)AL-D模型
南模生物長(zhǎng)期助力罕見病基因治療研究,構(gòu)建了Lipa-KO溶酶體酸性脂肪酶缺乏癥模型,助力溶酶體酸性脂肪酶缺乏癥相關(guān)機(jī)制研究需求,為相關(guān)藥物的藥效評(píng)估和安全性評(píng)價(jià)提供了強(qiáng)有力的工具。
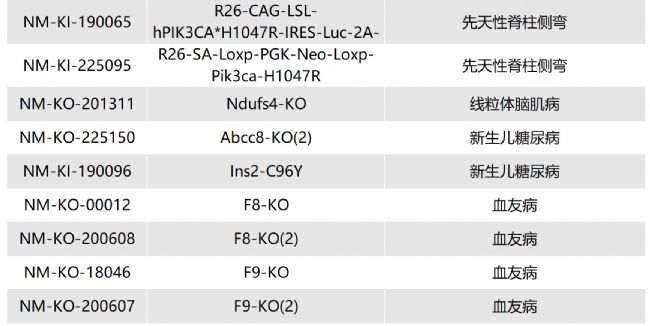
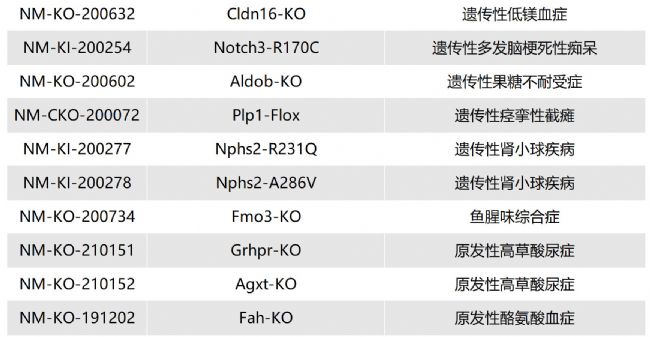
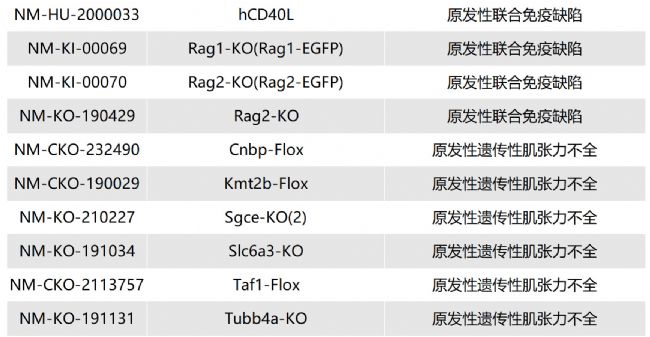
/ Lipa-KO(NM-KO-201298)/
部分驗(yàn)證數(shù)據(jù):
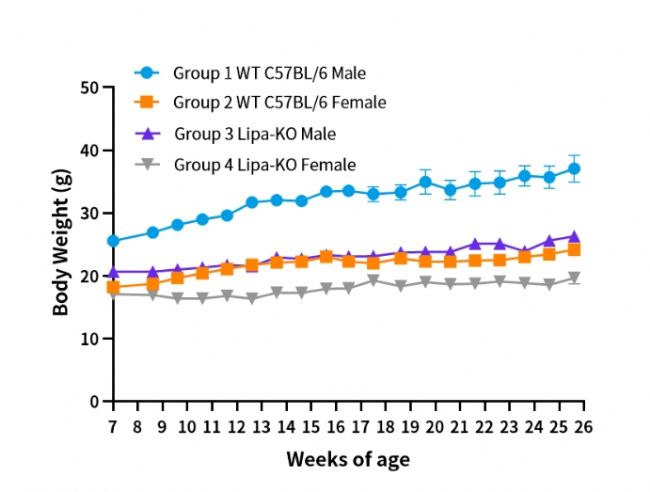
圖1. Lipa-KO小鼠體重變化。
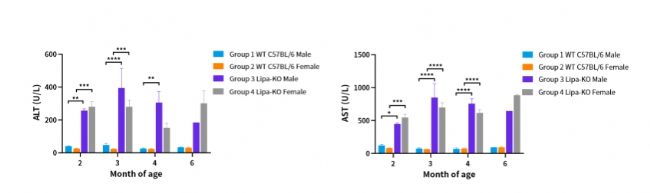
圖2. 不同月齡Lipa-KO小鼠血清中丙氨酸氨基轉(zhuǎn)移酶(alanine aminotransferase,ALT)和谷草轉(zhuǎn)氨酶(aspartate aminotransferase,AST)含量。
(2~4month: n=3~6; 6month: n=1~3)
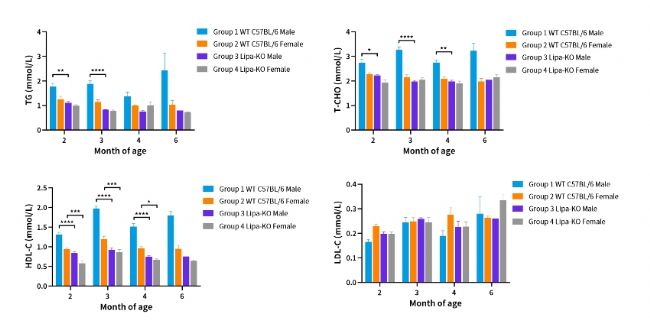
圖3. 不同月齡ipa-KO小鼠血脂分析。
(2~4month: n=3~6; 6month: n=1~3)
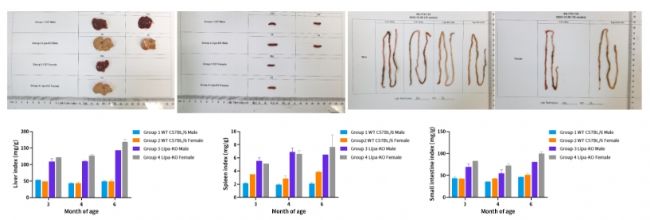
圖4. 肝臟中總膽固醇含量。(n=1~3)
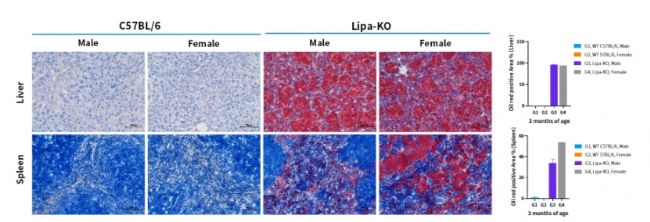
圖5. 3月齡Lipa-KO小鼠肝臟和脾臟的油紅染色代表性圖像及統(tǒng)計(jì)分析。
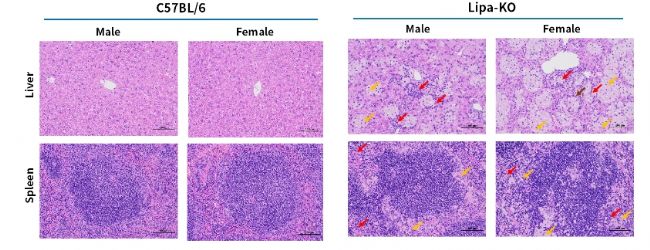
圖6. 3月齡Lipa-KO小鼠肝臟和脾臟的H&E染色代表性圖片。

如您有相關(guān)需求,歡迎撥打400-728-0660或者于微信公眾號(hào)南模生物SMOC在線咨詢,或致信marketing@modelorg.com,我們的專業(yè)團(tuán)隊(duì)將竭誠為您服務(wù)!
Reference:
[1] Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30. doi:10.1016/j.atherosclerosis.2014.04.003
關(guān)于我們
上海南方模式生物科技股份有限公司(Shanghai Model Organisms Center, Inc.,簡(jiǎn)稱"南模生物"),成立于2000年9月,是一家上交所科創(chuàng)板上市高科技生物公司(股票代碼:688265),始終以編輯基因、解碼生命為己任,專注于模式生物領(lǐng)域,打造了以基因修飾動(dòng)物模型研發(fā)為核心,涵蓋多物種模型構(gòu)建、飼養(yǎng)繁育、表型分析、藥物臨床前評(píng)價(jià)等多個(gè)技術(shù)平臺(tái),致力于為全球高校、科研院所、制藥企業(yè)等客戶提供全方位、一體化的基因修飾動(dòng)物模型產(chǎn)品解決方案。

- 譜系示蹤系列第三期之多重組酶介導(dǎo)的交叉報(bào)告基因方法
- 小鼠IVF-ET實(shí)驗(yàn)操作的流程與注意事項(xiàng)
- 溶酶體酸性脂肪酶缺乏癥(LAL-D)小鼠模型的介紹與應(yīng)用
- 原發(fā)性硬化性膽管炎(PSC)小鼠模型助力膽管炎的研究與治療
- 原發(fā)性膽汁性膽管炎(PBC)小鼠模型,為膽管炎研究按下加速鍵
- 慢性肺部疾病肺纖維化小鼠模型的介紹及應(yīng)用
- 從炎癥到疾病治療的關(guān)鍵角色白細(xì)胞介素IL-6簡(jiǎn)介及相關(guān)小鼠模型的應(yīng)用
- IL-23細(xì)胞因子誘導(dǎo)模型在助力銀屑病研究中的應(yīng)用
- 美國(guó)國(guó)立衛(wèi)生研究院宣布停止對(duì)僅動(dòng)物研究的資助
- 南模生物邀您共赴美國(guó)華人生物醫(yī)藥科技中國(guó)年會(huì)
- 集光講壇:10年基因編輯大牛講解Cre/loxP應(yīng)用全攻略
- 南模直播:一作解讀帕金森病治療新靶點(diǎn)FAM171A2
- 南模生物與GTP研發(fā)中心正式簽署GTP合作推廣計(jì)劃
- 明迅生物自主研發(fā)TIGIT人源化小鼠模型重磅來襲
- 南模生物將攜多個(gè)動(dòng)物模型最新研究成果亮相AACR年會(huì)
- 南模生物邀您相約華東第17屆實(shí)驗(yàn)動(dòng)物科學(xué)學(xué)術(shù)交流會(huì)